
Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
Aju lüssefalia
Artikli meditsiiniline ekspert

Viimati vaadatud: 12.07.2025
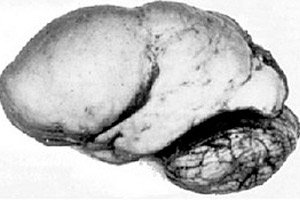
Orgaaniliste ajupatoloogiate hulgas paistab silma selline kaasasündinud aju arengu anomaalia nagu lissentsefaalia, mille olemus seisneb selle poolkerade koore peaaegu siledas pinnas - ebapiisava arvu keerdkäikude ja vagudega. [ 1 ]
Konvolutsioonide täielikul puudumisel defineeritakse agyriat ja mitmete laiade lamedate konvolutsioonide olemasolu nimetatakse pahügüüriaks. Need defektid, nagu mõned teised aju redutseeruvad deformatsioonid, kannavad ICD-10-s koodi Q04.3.
Epidemioloogia
Haruldaste haiguste statistika kohaselt on 100 tuhande vastsündinu kohta 1–1,2 lissentsefaalia juhtu. [ 2 ], [ 3 ]
Mõnede andmete kohaselt on Miller-Diekeri sündroomiga lastel täheldatud kuni 25–30% klassikalise lissentsefaalia juhtudest; LIS1 ja DCX geenide punktmutatsioone ja deletsioone tuvastatakse peaaegu 85%-l patsientidest. [ 4 ]
17 lissentsefaaliaga seotud geeni geneetilised uuringud näitasid, et LIS1 mutatsioon või deletsioon moodustab 40% patsientidest ja 23% on seotud DCX mutatsiooniga, millele järgnevad TUBA1A (5%) ja DYNC1H1 (3%).[ 5 ]
Põhjused Lissensefalia
Kõik teadaolevad ajukoore (cortex cerebri) peaaegu või täielikult ilma keerdude ja vagudeta moodustumise põhjused, mis suurendavad inimese aju "tööala" ja tagavad kesknärvisüsteemi "tootlikkuse", on seotud selle perinataalse arengu häiretega. See tähendab, et lootel tekib lissentsefaalia. [ 6 ]
Lissentsefaalia korral loote ajukoore kihtide moodustumise ebaõnnestumine on tingitud seda moodustavate neuronite ebanormaalsest migratsioonist või selle protsessi enneaegsest lõpetamisest.
See ajukoore histogeneesi jaoks hädavajalik protsess toimub mitmes etapis 7. kuni 18. rasedusnädalani. Ja arvestades selle suurenenud tundlikkust geneetiliste mutatsioonide, aga ka mitmesuguste negatiivsete füüsikaliste, keemiliste ja bioloogiliste mõjude suhtes, võib igasugune normist kõrvalekalle viia neuronite vale lokaliseerimiseni, mille tulemuseks võib olla ajukoore halli aine paksenenud kihi moodustumine ilma iseloomuliku struktuurita. [ 7 ]
Mõnel juhul on lastel esinev lissentsefaalia seotud Miller-Diekeri, Walker-Warburgi või Norman-Roberti sündroomidega.
Loe ka – Aju arenguhäired
Riskitegurid
Lisaks mutatsioonidele mõnedes geenides on sellise raske defektiga lapse sünni riskiteguriteks loote hapnikupuudus (hüpoksia); ebapiisav aju verevarustus (hüpoperfusioon); äge tserebrovaskulaarne õnnetus perinataalse insuldi kujul; platsenta patoloogiad; raseda naise viirusnakkused (sh TORCH); [ 8 ] üldise ainevahetuse ja kilpnäärme talitluse probleemid; suitsetamine, alkohol, psühhotroopsed ja narkootilised ained; mitmete ravimite tarvitamine; suurenenud kiirgustase. [ 9 ]
Pathogenesis
Mitte kõigil lissentsefaalia juhtudel ei ole patogeneesi põhjustanud kromosoomanomaaliad ja geenimutatsioonid. Siiski on teada mõned geenid, mis kodeerivad valke, millel on oluline roll neuroblastide ja neuronite korrektses liikumises mööda radiaalseid gliarakke – et moodustada ajukoor. Ja nende geenide mutatsioonid viivad selle patoloogiani. [ 10 ]
Täpsemalt öeldes on need LIS1 geeni juhuslikud mutatsioonid (ilma pärilikkuseta) 17. kromosoomis, mis reguleerib mikrotuubulite tsütoplasmaatilist motoorvalku düneiini, samuti DCX geeni X-kromosoomis, mis kodeerib valku doublekortiini (lissentsefaliin-X). [ 11 ]. Esimesel juhul defineerivad spetsialistid klassikalist lissentsefaaliat (I tüüp), teisel X-seotud. [ 12 ]
Kui FLN1 geen, mis kodeerib fosfoproteiini filamiin 1, on kustutatud, ei pruugi suunatud neuronite migratsiooni protsess üldse alata, mis viib konvolutsioonide (agyria) täieliku puudumiseni. [ 13 ]
CDK5 geenis, mis kodeerib kinaasi ensüümi – rakusisese metabolismi katalüsaatorit, reguleerib kesknärvisüsteemi neuronite rakutsüklit ja tagab nende normaalse migratsiooni aju struktuuride sünnieelse moodustumise ajal, on tuvastatud mutatsioonid.
Norman-Robertsi sündroomi korral 7. kromosoomis asuva RELN geeni ebanormaalsed muutused, mis põhjustavad kortikaalseid güraalseid defekte, põhjustavad rakuvälise glükoproteiini reeliini defitsiiti, mis on vajalik neuraalsete tüvirakkude migratsiooni ja positsioneerimise reguleerimiseks ajukoore arengu ajal.[ 14 ], [ 15 ], [ 16 ]
ARX geen kodeerib mitte-aristalensi homeoboxvalku, transkriptsioonifaktorit, millel on oluline roll eesajus ja teistes kudedes.[ 17 ] ARX mutatsiooniga lastel esinevad ka muud sümptomid, näiteks puuduvad ajuosad (corpus callosumi agenees), ebanormaalsed suguelundid ja raske epilepsia.[ 18 ],[ 19 ]
Lissentsefaaliaga on seostatud mitmeid geene. Nende geenide hulka kuuluvad VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A ja CDK5.[ 20 ]
Tsütomegaloviirust (CMV) on seostatud lissentsefaalia tekkega, mis on tingitud loote aju verevarustuse vähenemisest. CMV-nakkuse raskusaste sõltub raseduse vanusest. Varajane infektsioon põhjustab tõenäolisemalt lissentsefaaliat, kuna neuronite migratsioon toimub raseduse alguses.[ 21 ]
Lisaks hõlmab selle anomaalia esinemise mehhanism neuronite migratsiooni mittetäielikku või hilisemat peatumist periventrikulaarsest generatiivsest tsoonist ajukoorde. Sellistel juhtudel tekib kas mittetäielik lissentsefaalia või pahügüüria, mille käigus moodustuvad mitu laia soont ja konvolutsiooni (kuid enamik neist puudub).
Sümptomid Lissensefalia
Selle patoloogia esimesed tunnused (eelnevalt mainitud sündroomide puudumisel) ei pruugi ilmneda kohe pärast sündi, vaid poolteist kuni kaks kuud hiljem. Ja enamasti täheldatakse järgmisi lissentsefaalia kliinilisi sümptomeid:
- lihashüpotoonia, sageli koos spastilise halvatusega;
- krambid ja üldised toonilis-kloonilised krambid (opistotoonuse kujul);
- raske vaimne alaareng ja kasvupeetus;
- neuroloogiliste ja motoorsete funktsioonide kahjustus.
Neelamisprobleemid raskendavad lapse toitmist. [ 22 ]
Raske neuromotoorse kahjustuse korral avaldub sageli tetrapleegia – kõigi jäsemete halvatus. Võimalik on käte, sõrmede või varvaste deformatsioon.
Norman-Roberti sündroomi ja I tüüpi lissentsefaalia korral täheldatakse kraniofakiaalseid anomaaliaid: rasket mikrotsefaaliat, madalat laubajoont ja väljaulatuvat laia ninaselga, laia asetusega silmad (hüperterlorism), lõualuude arengupeetust (mikrognaatiat). [ 23 ]
Miller-Diekeri sündroomi võib iseloomustada ka ebanormaalselt väike pea suurus laia ja kõrge lauba ning lühikese ninaga, lohud oimukohtades (bitemporaalsed lohud) ja madalal asetsevad deformeerunud kõrvad.
Raskekujulist lissentsefaalia sündroomi iseloomustab mikrotsefaalia ehk silmamunade suuruse vähenemine (mikroftalmia) koos võrkkesta düsplaasia, obstruktiivse hüdrotsefaalia ja puuduva või hüpoplastilise mõhnkehaga.
Tüsistused ja tagajärjed
Selle anomaalia tüsistuste hulgas nimetavad spetsialistid neelamisfunktsiooni (düsfaagia) ja gastroösofageaalse refluksi rikkumist; refraktaarset (kontrollimatut) epilepsiat; sagedasi ülemiste hingamisteede infektsioone; kopsupõletikku (sh kroonilist aspiratsiooni).
Lissentsefaaliaga imikutel võivad esineda kaasasündinud orgaanilised südameprobleemid kodade vaheseina defekti või keerulise südamerikke kujul koos tsüanoosiga (Fallot' tetraloogia). [ 24 ]
Sünnitusjärgse kasvupeetuse tagajärjed lõppevad enamasti surmaga 24 kuu jooksul pärast sündi.
Diagnostika Lissensefalia
Diagnoos algab lapse füüsilise läbivaatuse, vanemate haigusloo ning raseduse ja sünnituse ajaloo uurimisega.
Raseduse ajal võib olla vajalik loote DNA-test ilma rakkudeta, amniotsentees või koorionivilla proovide võtmine. [ 25 ] Lisateabe saamiseks vt – Kaasasündinud haiguste sünnieelne diagnoosimine
Instrumentaalset diagnostikat kasutatakse aju struktuuride visualiseerimiseks ja nende funktsioonide hindamiseks:
- aju kompuutertomograafia;
- aju magnetresonantstomograafia (MRI);
- elektroentsefalogramm (EEG). [ 26 ]
Raseduse ajal võib loote ultraheliuuringul 20–21 nädala pärast kahtlustada lissentsefaaliat parieto-oktsipitaalsete ja kaltsariiniliste vagude puudumisel ning aju Sylvia lõhe anomaalia korral.
Diferentseeritud diagnoos
Teostatakse diferentsiaaldiagnostika teiste kaasasündinud ajukahjustuste sündroomidega.
Lissentsefaaliat on üle 20 tüübi, millest enamik jaguneb kahte põhikategooriasse: klassikaline lissentsefaalia (1. tüüp) ja munakiviliissentsefaalia (2. tüüp). Igal kategoorial on sarnased kliinilised ilmingud, kuid erinevad geneetilised mutatsioonid.[ 27 ]
I tüüpi lissentsefaalia korral tehtud aju-uuring näitab nelja kihiga ajukoore kuue kihi asemel, mis on näha tervetel patsientidel, samas kui II tüüpi lissentsefaalia korral on ajukoor korrastamata ja tundub tükiline või sõlmeline, kuna ajukoore on täielikult nihkunud ajukoore neuronite klastrite poolt, mida eraldab gliomeenhümaalne kude. Patsientidel esines ka lihaste ja silmade kõrvalekaldeid.
- Klassikaline lissentsefaalia (tüüp 1):
- LIS1: Isoleeritud lissentsefaalia ja Miller-Diekeri sündroom (näo düsmorfismiga seotud lissentsefaalia). [ 28 ]
- LISX1: DCX geeni mutatsioon. Võrreldes LIS1 mutatsioonide põhjustatud lissentsefaaliaga on DCX-il nelja asemel kuuekihiline ajukoor.
- Isoleeritud lissentsefaalia ilma muude teadaolevate geneetiliste defektideta
- Munakivist tingitud lissentsefaalia (tüüp 2):
- Walker-Warburgi sündroom
- Fukuyama sündroom
- Lihaste, silmade ja aju haigused
- Teisi tüüpe ei saa paigutada ühte kahest ülaltoodud rühmast:
- LIS2: Norman-Roberti sündroom, sarnane I tüüpi lissentsefaalia või Miller-Diekeri sündroomiga, kuid ilma 17. kromosoomi deletsioonita.
- LIS3
- LISX2
Mikrolissentsefaalia: see on kombinatsioon normaalse ajukoore voltimise puudumisest ja ebanormaalselt väikesest peast. Tavapärase lissentsefaaliaga lastel on sündides normaalse suurusega pea. Väikese pea suurusega sündides diagnoositakse tavaliselt mikrolissentsefaalia.
Samuti on oluline eristada lissentsefaaliat ja polümikrogüüriat, mis on erinevad aju arenguhäired.
Kellega ühendust võtta?
Ravi Lissensefalia
Lissentsefaalia on ravimatu orgaaniline defekt, seega on võimalik ainult toetav ja sümptomaatiline ravi. [ 29 ]
Esiteks on see krambivastaste ja epilepsiavastaste ravimite kasutamine, samuti gastrostooma paigaldamine maosse (kui laps ei suuda iseseisvalt neelata). Massaaž on kasulik.
Raske hüdrotsefaali korral eemaldatakse tserebrospinaalvedelik.
Ärahoidmine
Eksperdid soovitavad tulevastel vanematel pöörduda geneetilise nõustamise poole ning rasedatel naistel registreeruda õigeaegselt günekoloogide ja sünnitusarstide vastuvõtule ning läbida kõik plaanilised uuringud.
Prognoos
Lissentsefaaliaga laste puhul sõltub prognoos selle astmest, kuid enamasti ei ületa lapse vaimne areng nelja kuni viie kuu piiri. Ja kõik selle diagnoosiga lapsed kannatavad raskete psühhomotoorsete häirete ja raskesti ravitava epilepsia all. [ 30 ]
NINDS-i (Ameerika Riiklik Neuroloogiliste Häirete ja Insuldi Instituut) andmetel on lissentsefaaliaga inimeste maksimaalne eluiga umbes 10 aastat.